







How about going next with us?
For more information, please feel free to contact us from our inquiry
form!!
GRRM23
Automatic chemistry program for exploring all reaction paths and molecules
Global reaction route mapping (GRRM) program is a quite powerful calculation tool
developed to pave the way for widespread exploration of chemical reaction paths by working with quantum
chemistry calculation engines (like Gaussian and others).
What makes GRRM differ from the other
software is that, throughout its automatic reaction path search starting from just one single input of
molecule data, this program enables users to find out all possible chemical products, transition states,
and reaction yeilds comprehensively.
GRRM continues to inspire researchers & engineers to accelearate
their R&D in the various fields related to chemistry including catalyst design and material screening,
and has now evolved to the latest version with a new advanced retrosynthetic analysis function as
"GRRM23".
- 01
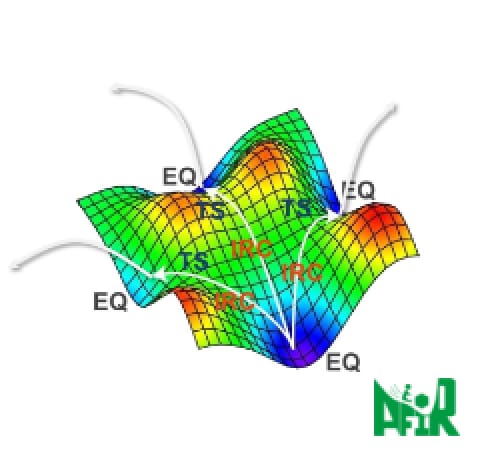
AFIR and ADDF algorithm for automatic exploration of all potential molecules and reaction paths
Global reaction route mapping (GRRM) program empowers users to do their treasure hunt of unknown molecules and reactions through automatic reaction path exploration from one starting point (equilibrium), driven by two core algorithms that collaborate with quantum chemistry calculation: artificial force induced reaction (AFIR) and anharmonic downward distortion following (ADDF). The power of ADDF and AFIR opens unknown chemistry of all four types of chemical reactions shown below [1], which leads users to explore possible output (molecules and reaction paths) comprehensively just by one single input of molecules.
(1) A → X (Isomerization)
(2) A → X+Y (Dissociation)
(3) A+B → X (Combining synthesis)
(4) A+B → X+Y (Exchanging synthesis) - 02
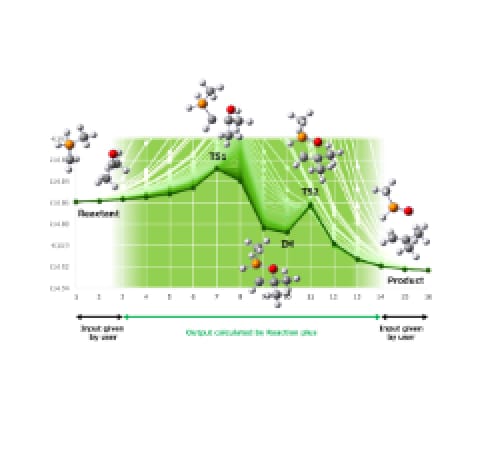
Kinetic analysis by RCMC method to evaluate population of chemical species and accelerate reaction path search
Kinetic simulation and kinetics-based navigation using rate constant matrix contraction (RCMC) method became available from GRRM20 to enhance the automated reaction search function of GRRM, being applicable to complex reaction path networks. The kinetic simulation enables users to evaluate population of chemical species which exist beyond specified lifetime condition, while the kinetics-based navigation contributes to dramatic speedups of the reaction path search (SC-AFIR search of GRRM) by restricting the search areas to extract kinetically feasible paths from initial structures under given reaction temperatures and lifetimes [2].
- 03
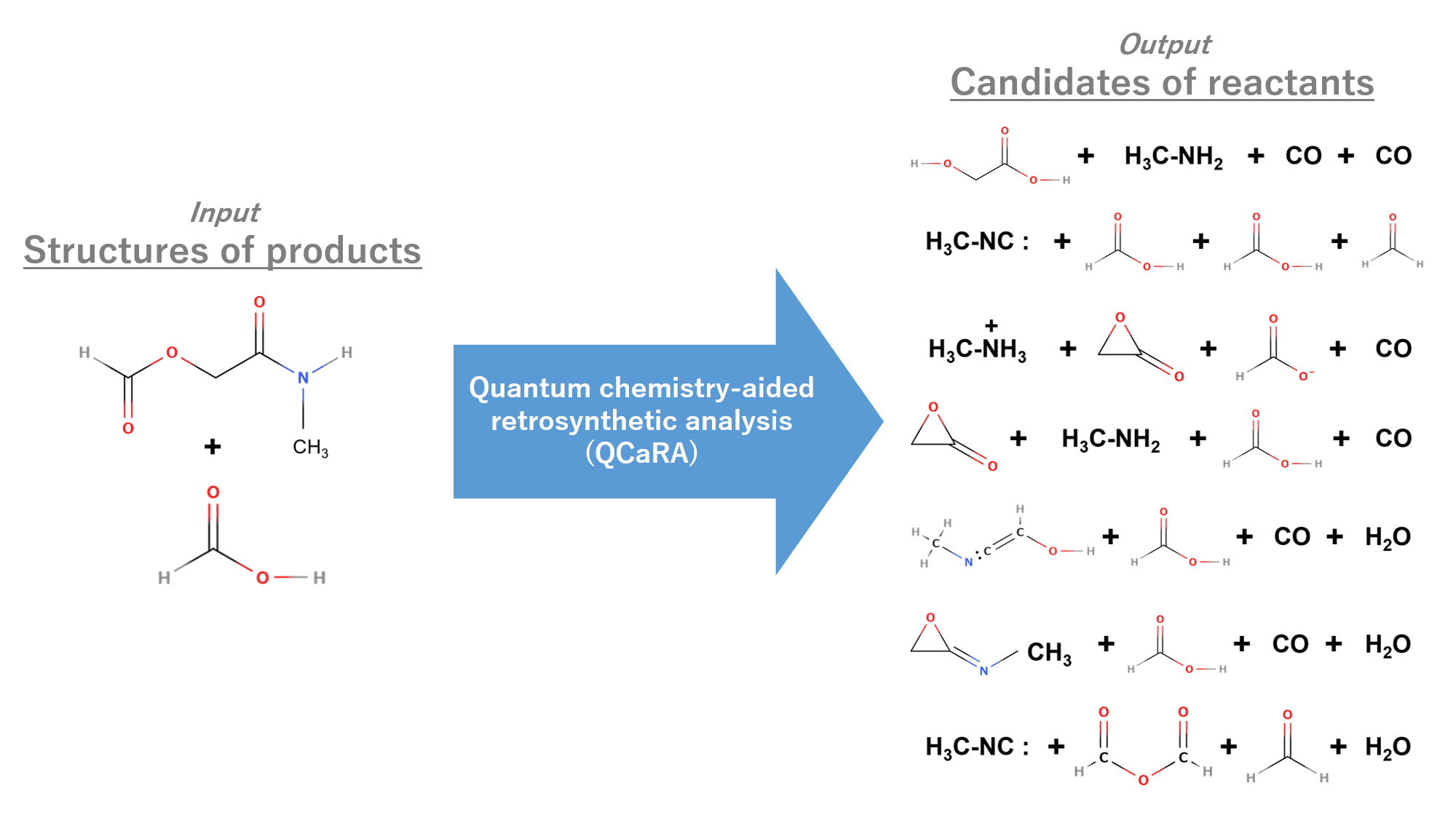
Retrosynthetic analysis for exploring chemical reactants and yields by QCaRA
In GRRM23, the new function for reverse reaction kinetics analysis was implemented based on RCMC method (above-mentioned) for complex reaction path network analysis. This function predicts yields of target chemical products regarding all reactions starting from other chemical species on reaction path networks [3]. By utilizing this function as a kinetics navigation tool, quantum chemistry-aided retrosynthetic analysis (QCaRA) is performed. So far, QCaRA, which takes structures of products as its sole input, has already demonstrated the ability to correctly identify reactants for various known reactions, including the synthesis of small natural products [4].
- 04
Utilities to easily implement user-developed tools
GRRM23 provides functions to control structure optimization and exploration using externally developed modules. These modules can modify search order near local minima, change exploration routes from local minima, and apply external bias potentials to the system. These options are used in combination with rapidly exploring random tree algorithm [5] and graph neural network-based reaction path selection algorithm [6] in order to accelerate the reaction path exploration (SC-AFIR search of GRRM) for specific purposes. Moreover, they are also used for the development and application of virtual ligands for transition metal catalysts [7].
- 05
Applicable for various R&D
GRRM23 can be used to simulate various reaction systems, so far having already been applied in organic reaction, organometallic catalysis, cluster catalysis, radical reaction, photoreaction involving electronic excited states, crystal phase transition under periodic boundary conditions, enzyme catalysis reaction by QM/MM-ONIOM method, etc.
For example, in drug synthesis, GRRM program can be utilized for catalyst design targeting rate-determining reactions and for suppression of by-products by elucidating all reaction paths capable of synthesizing desired drug molecules.
For example, in combustion reaction, it is possible to meet the accuracy of reaction rates in CAE design of automotive and rocket engine because thousands to tens of thousands of elementary reactions can be obtained based on highly precise quantum chemistry calculations.
Visit developer's site
for more info
See AFIR manual site
See AFIR manual site
Calculation examples
| Input | Output | |
|---|---|---|
| Number of stable structures (Reactants / Products / Intermediates) | Number of elementary reactions | |
| acetic acid CH3COOH | 121 | 848 |
| propionic acid C3H2O2 | 207 | 1,114 |
| Methyl nitrate CH3NO3 | 676 | 4,835 |
| Lactaldehyde C3H6O2 | 1,366 | 10,103 |
Major publications
- A Scaled Hypersphere Search Method for the Topography of Reaction Pathways on the Potential Energy Surface. K. Ohno and S. Maeda Chem. Phys. Lett., 384(4-6), 277-282 (2004).
- A Scaled Hypersphere Search Method for the Topography of Reaction Pathways on the Potential Energy
Surface. K. Ohno and S. Maeda Chem. Phys. Lett., 384(4-6), 277-282 (2004).
Systematic Exploration of the Mechanism of Chemical Reactions: Global Reaction Route Mapping (GRRM) Strategy by the ADDF and AFIR Methods. Satoshi Maeda, Koichi Ohno, and Keiji Morokuma Phys. Chem. Chem. Phys., 15, 3683-3701 (2013). - Global Mapping of Equilibrium and Transition Structures on Potential Energy Surfaces by the Scaled Hypersphere Search Method: Application to ab initio Surfaces of Formaldehyde and Propyne Molecules. S. Maeda and K. Ohno J. Phys. Chem. A 109(25), 5742-5753 (2005).
Visit developers' sites for more info
Non-functional specs
- Item
- Content
- Operating environment
-
- Hardware
x86_64 computer that works below - OS
Red Hat Enterprise Linux 7.x or CentOS 7.x
Red Hat Enterprise Linux 8.x, CentOS 8.x or AlmaLinux 8.x
(Red Hat Enterprise Linux 6.x and CentOS 6.x are not supported.) - Required software
Gaussian16 or Gaussian09 *1 - Optional software
Gaussian03, Molpro, GAMESS, ORCA, Turbomole, SIESTA *2 *3
- Hardware
- Included items
- Software *4
*1 Not included in this product. Please prepare in advance.
*2 Not included in this product. Please prepare in advance as needed.
*3 A general interface to insert ab initio programs besides these is contained in GRRM23.
*4 Please refer to AFIR manual site for the latest GRRM23 info.
Functional specs
Update functions
- Function
- GRRM20
- GRRM23
- Quantum chemistry-aided retrosynthetic analysis (QCaRA)
- ✕
- ○
- Utilities to easily implement user-developed tools
- ✕
- ○
See more
See more
GRRM23
GET IN TOUCH
GET IN TOUCH
For more information, please contact us.
We support users to set up and start GRRM23 (free of charge in standard).
As for the further technical information such as how to use and how to interpret results, please note in advance that users are encouraged to inquire through AFIR forum site (registration on AFIR site is required when purchasing GRRM23 license).
Please refer to AFIR manual site for the latest manual.
- November 6th, 2023
-
Released "GRRM23"!!
May 19th, 2023