







Table of contents
- 1. Background
- 1-1. Why computational chemistry?
- 1-2. Advanced chemistry program: GRRM
- 2. Our works
- 2-1. Challenges to boost GRRM calculation with supercomputer Fugaku
- 2-2. Achievement by Fugaku-GRRM integrated system
- 2-3. Future perspective
1-1.Why computational chemistry?
All the substances in our everyday life are comprised of the combination of chemical elements and the
discoveries of new chemical compounds have played a crucial role in development of various industries to
shape our world. For instance, cross-coupling reactions are estimated to have an industrial impact of
approximately 60 trillion yen in pharmaceuticals, liquid crystal displays, light-emitting diodes (LEDs),
etc.
For these reasons, chemists are striving for innovations through their R&D every day.
While most of their works has been done by mainly experimental methods, the importance of simulation
technologies by computational chemistry has been increasing to elucidate unknown chemical compounds and
mechanisms that leads to acceleration of development. Therefore, various kinds of computational
chemistry software have been developed these days to support their investigation.
1-2.Advanced chemistry program: GRRM
In the field of quantum chemistry where prediction of nano-scale molecules is carried out as a part of
computational chemistry, scientists have been pursuing a dreamlike way that can automatically and
comprehensively create a world map of chemical reaction network from a single molecular data. This
approach is important for them to unearth treasures of information beneficial for synthesizing new
chemical substances, but has been extremely difficult just by conventional quantum chemistry
software.
In order to overcome this problem, an innovative program “GRRM”*1has been
developed by Prof. Maeda’ s research group*2to enhance the quantum chemistry software. Driven
by powerful AFIR*3algorithm inside the program, GRRM paves the way for widespread automatic
exploration of chemical reactions, which leads researchers to find out unknown molecular structures and
reaction mechanism. Owing to its capability, GRRM already has a proven track record of applications in
various reaction systems, including organic reactions, catalysis reactions, photochemical reactions, and
phase transitions in crystals and others.
Although GRRM is gaining attention worldwide as a promising
R&D tool especially among computational chemists, its substantial calculation cost is a major
hurdle for practical use. This is because it requires a vast amount of quantum chemistry calculations to
explore the expansive multidimensional space of chemical reaction paths (3N-6 dimensions where N is the
number of atoms), leading to fatally long calculation time for industrial development.
*1 GRRM (Global Reaction Route Mapping):
Automatic calculation program
capable of exploring all possible chemical reactions as well as molecular structures from one molecule
data through its global reaction path search in conjunction with quantum chemistry calculation software
(like Gaussian).
HPC SYSTEMS is the provider of this program.
https://global.hpc.co.jp/products/grrm23/
*2 Prof. Maeda’s research group:
Theoretical Chemistry lab, Frontier
Chemistry Center, Hokkaido University.
The group is at the forefront in development of GRRM program
and AFIR algorithm.
https://sites.google.com/site/grrmgroup/home
*3 AFIR (Artificial Force Induced Reaction):
Core algorithm
implemented in GRRM. It is designed to promote chemical reactions in calculations by relieving energy
barriers artificially, playing an essential role in reaction path search of GRRM.
https://afir.sci.hokudai.ac.jp/
2-1.Challenges to boost GRRM calculation with supercomputer Fugaku
One of the effective solutions to the calculation cost for GRRM simulation, is to allocate abundant CPU
resources and execute explorations of multiple chemical reaction paths in parallel. This approach
thereby accelerates computation speed and reduces calculation time for GRRM’s extensive exploration
system. To achieve this, there has been growing interest in leveraging supercomputers equipped with vast
CPU resources.
In response to this challenge, HPC SYSTEMS Inc. has been collaborating with Prof.
Maeda’s research group. This joint project aims to harness the power of RIKEN's supercomputer
"Fugaku"*4to enhance the capabilities of GRRM driven by AFIR algorithm. The
objective has been to facilitate the large-scale automated exploration of chemical reactions in a short
period by integrating GRRM program with “A64FX”*5processor of Fugaku. In this project,
attempts have been made to fine-tune the codes of GRRM and others for seamless calculation on A64FX
processor. As a system integrator in the project, HPC SYSTEMS has contributed to optimization of
GRRM-AFIR program for the system architecture of Fugaku (Figure 1).
Subsequently, evaluations have
been conducted to verify its performance and feasibility.
*4 RIKEN (Institute of Physical and Chemical Research) and
Fugaku:
RIKEN is one of the prominent national scientific research institutes and it has the
Japan-made supercomputer Fugaku.
https://www.r-ccs.riken.jp/en/fugaku/
*5 A64FX:
The world's first processor inducing Scalable Vector
Extension (SVE), an extension of the Armv8.2-A instruction set architecture for supercomputers,
developed by Fujitsu Limited.
https://www.fujitsu.com/global/products/computing/servers/supercomputer/a64fx/
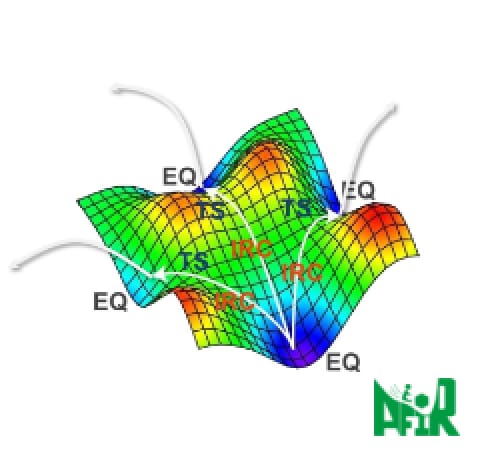
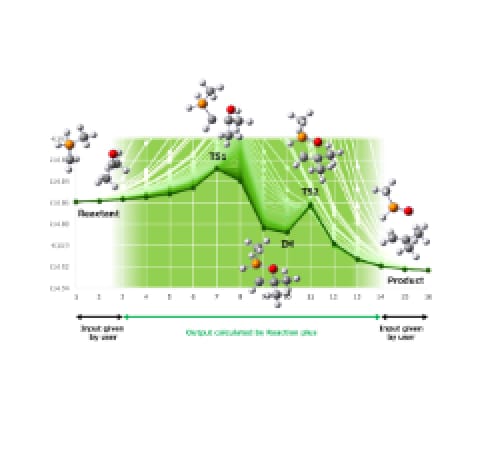
 Figure 1HPC SYSTEMS has contributed to optimization
of GRRM-AFIR program for the supercomputer Fugaku
Figure 1HPC SYSTEMS has contributed to optimization
of GRRM-AFIR program for the supercomputer Fugaku
2-2.Achievement by Fugaku-GRRM integrated system
By leveraging the immense computing power of Fugaku, a significant advancement has been made in the
large-scale simulation of chemical reaction using GRRM *6. Starting from February 1, 2021, we
concurrently harnessed up to 200,000 CPU cores of Fugaku for our calculations. As a result, this led to
the exploration of approximately 930,000 reaction paths and around 120,000 chemical compounds within
just 50 days (Figure 2).
Our success is attributed to our cumulative efforts of system integration,
including implementation of quantum chemistry software “Gaussian”*7(which is called from
GRRM) on Fugaku and optimization of file location by utilizing LLIO*8high-speed file system
to ensure efficient and stable operation of GRRM on Fugaku.
In summary, this accomplishment showcases
the extraordinary computational capabilities of Fugaku through tailored integration by HPC SYSTEMS.
*6 T. Mita, H. Takano, H. Hayashi, W. Kanna, Y. Harabuchi, K. N. Houk, S. Maeda, "Prediction of High-Yielding Single-Step or Cascade Pericyclic Reactions for the Synthesis of Complex Synthetic Targets", J. Am. Chem. Soc., 2022, 144, 50, 22985–23000. DOI:10.1021/jacs.2c09830
*7 Gaussian:
De facto standard software in the field of quantum
chemistry.
It is used as the quantum chemistry calculation engine in GRRM simulation.
https://gaussian.com/
*8 LLIO (The Lightweight Layered IO-Accelerator):
The dedicated file
system adopted in Fugaku's job execution area, developed for improving hierarchical storage
usability and optimizing application file I/O based on file-use characteristics.
https://www.fujitsu.com/global/about/resources/publications/technicalreview/2020-03/article05.html
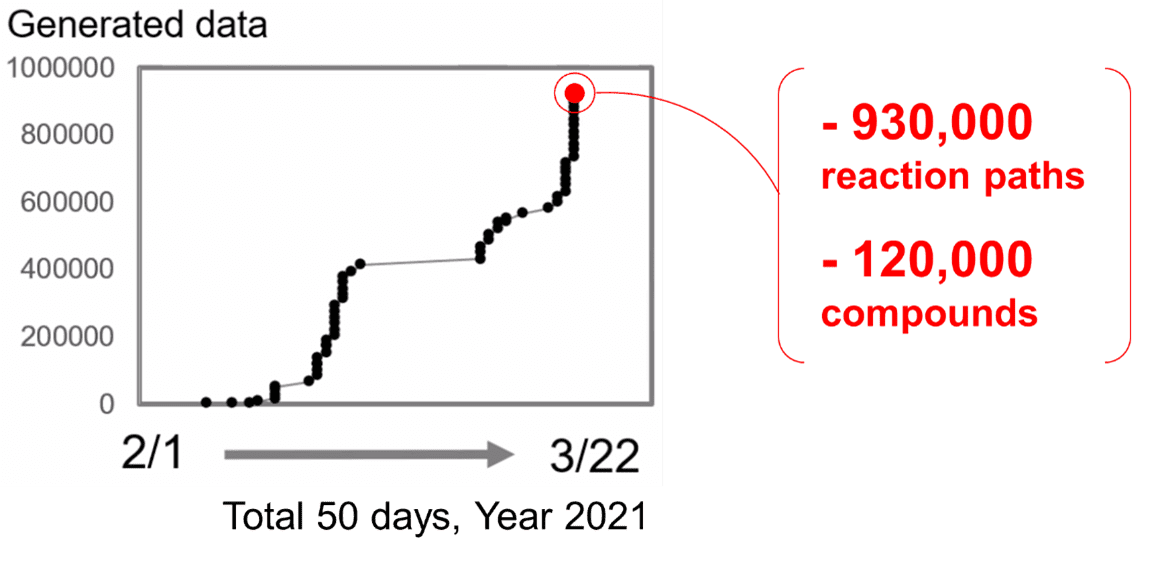 Figure 2Accumulative number of chemical reaction
paths found by GRRM on Fugaku
Figure 2Accumulative number of chemical reaction
paths found by GRRM on Fugaku
2-3.Future perspective
Our project of Fugaku-GRRM system is motivated to pioneer the domain of large-scale automatic
explorations of chemical reactions, potentially leading to groundbreaking discoveries that brings a big
impact to industries. By applying the abundant data generated from this system to fields such as
materials informatics*9and synthetic robotics,*10especially where vast datasets
are exploited for materials development, industrial advancements can be accelerated
further.
Acknowledging this potential, proactive measures are undertaken to expand user access by
offering the system resources through HPC SYSTEMS’s cloud platform “Science Cloud”*11and a
RIKEN-leaded project for chemistry application users. These approaches aim to facilitate widespread
utilization as well as development and verification of the system for the future industry.
*9 Materials informatics:
The interdisciplinary field that uses
computational methods, data science, and machine learning to analyze and extract valuable insights from
vast datasets related to materials, enabling the discovery and optimization of new materials for various
applications.
*10 Synthetic robotics:
The cutting-edge field involving integration
of automation, robotics, and advanced algorithms to facilitate and optimize chemical synthesis
processes. These robotic systems can automate complex synthesis tasks, enhance precision, and increase
throughput in chemical production, streamlining the development of new compounds and formulations.
*11 Science Cloud:
The original cloud services of HPC SYSTEMS,
providing the cloud platform that includes HPC environment with pre-installed science software.
https://global.hpc.co.jp/products/science-cloud/