







How about going next with us?
For more information, please feel free to contact us from our inquiry
form!!
Chemical Reaction Software
GRRM26
Comprehensive, automatic exploration of chemical reaction networks
The Global Reaction Route Mapping (GRRM) program is a powerful calculation tool
developed to open up broad exploration of chemical reaction pathways, working together with quantum
chemistry calculations and machine learning potentials.
What sets GRRM apart is that, through an
automatic reaction pathway search starting from just a single molecular input, it lets users
comprehensively map out reaction pathway networks — revealing chemical products, transition states, and
reaction yields that can emerge along the way.
GRRM continues to inspire researchers & engineers
to accelerate their R&D across many fields of chemistry, from catalyst design to materials screening
— and it keeps evolving, with the latest version, "GRRM26", adding a range of new functions for
comprehensive exploration.
- 01
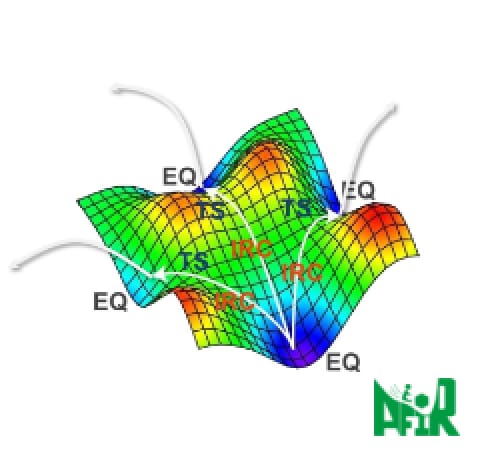
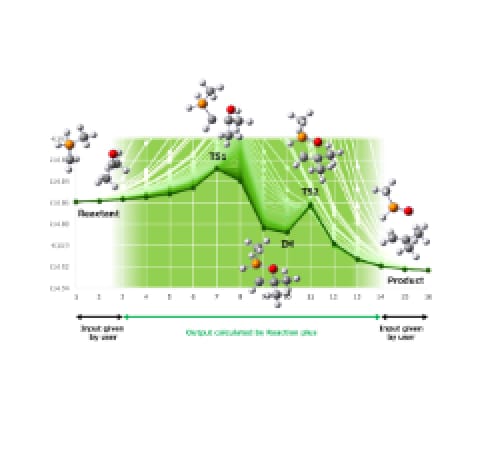
Comprehensive reaction pathway exploration — AFIR
GRRM lets users go on a treasure hunt for unknown molecules and reactions: from a single starting structure (an equilibrium geometry), it automatically explores reaction pathways by working together with quantum chemistry calculations and machine learning potentials (MLP). GRRM's development began with the anharmonic downward distortion following (ADDF) method, and its exploration is now driven mainly by the artificial force induced reaction (AFIR) method — GRRM's current core search algorithm. AFIR and ADDF are two independent methods, and together they uncover all four fundamental types of chemical reaction [1], so that a single molecular input can map out the possible products and reaction pathways comprehensively.
(1) A → X (Isomerization)
(2) A → X + Y (Dissociation)
(3) A + B → X (Combining synthesis)
(4) A + B → X + Y (Exchanging synthesis)New!! Optimization of search performance for high-speed energy calculation programs.
Search performance has been significantly improved when using fast energy and gradient evaluation methods such as semi-empirical methods (SEM) and machine learning potentials (MLP). It is now possible to routinely construct reaction pathway networks containing more than 1,000 pathways using fast methods like SEM and MLP [2][3][4].
- 02
Kinetic simulation and kinetics-based navigation — RCMC
Kinetic simulation and kinetics-based navigation using the rate constant matrix contraction (RCMC) method — available since GRRM20 — extend GRRM's automated search to complex reaction pathway networks. The kinetic simulation evaluates the population of chemical species that persist beyond a specified lifetime, while the kinetics-based navigation dramatically speeds up the reaction pathway search (SC-AFIR) by restricting it to kinetically feasible paths under given reaction temperatures and lifetimes [5].
New!! Explicit consideration of concentration effects in RCMC calculations.
Prior to GRRM23, reaction pathway network representations were limited to stoichiometric reactions where all reagents were mixed in equal proportions (the molar ratio of all reagents being 1:1:1…) due to limitations in the computational algorithm. The new algorithm introduced in GRRM26 enables RCMC kinetic simulations that explicitly consider concentration effects, making it possible to apply them to complex systems where catalyst coating rate and reagent concentration affect reaction efficiency [6].
New!! RCMC under a Microcanonical Ensemble condition.
It is now possible to perform RCMC kinetic simulations under a Microcanonical Ensemble. The implemented full-RCMC (f-RCMC) method enables the calculation of final branching ratios for products without performing time evolution of the rate equations. While conventional methods take approximately 95 hours to solve the rate equation for C4H5, consisting of 234 elementary processes, f-RCMC has been reported to calculate the branching ratio in less than 1 second [7].
[6]S. Maeda, et al. (to be published) - 03
Retrosynthetic analysis — QCaRA
Building on the RCMC method, GRRM performs reverse (retrosynthetic) kinetic analysis across complex reaction pathway networks. It predicts the yields of a target product for all reactions that could lead to it from other species on the network [8]. Used as a kinetics navigation tool, this enables quantum chemistry-aided retrosynthetic analysis (QCaRA): taking only the structure of a product as its input, QCaRA has already correctly identified the reactants of various known reactions, including the synthesis of small natural products [9].
- 04
External interface integration
GRRM works together with a range of external computation engines — most notably Gaussian, along with ORCA, GAMESS and others — and, through its general external interface, it is designed to make it easy to connect other solvers that users already have, whether quantum chemistry engines or machine learning potentials (MLP), beyond the officially supported ones. Building on this interface, GRRM can also control structure optimization and exploration with externally developed modules — modifying the search order near local minima, redirecting exploration routes, or applying external bias potentials — and it is the same interface that lets GRRM couple with machine learning potentials. Combined with a rapidly exploring random tree (RRT) algorithm [10] and a graph neural network for path selection [11], it accelerates the reaction pathway search (SC-AFIR) for specific purposes, and it underpins the development and application of virtual ligands for transition-metal catalysts [12].
New!! Improved adaptability to the virtual ligand method and related techniques.
By integrating with the virtual ligand method and its code developed by Specially Appointed Associate Professor Wataru Matsuoka and his colleagues, ligand optimization of organometallic catalysts can be performed. References [13] and source code [14] for the virtual ligand method are provided below.
- 05
And many more functions
Beyond the highlights above, GRRM has steadily accumulated a wide range of functions for comprehensive reaction pathway exploration. For a full, side-by-side view of what each version supports, see the comparison table under Functional specs ("See more").
New!! Other performance improvements.
The GRRM source code has been completely redesigned, and numerous performance improvements have been implemented.
GRRM is the program developed by the research group of Professor Satoshi Maeda at Hokkaido University, and HPC SYSTEMS provides its license as a partner. For more technical information, visit the developer's site.
See AFIR-web
See AFIR-web
Calculation examples
| Input | Output | |
|---|---|---|
| Number of stable structures (Reactants / Products / Intermediates) | Number of elementary reactions | |
| Acetic acid CH3COOH | 121 | 848 |
| Propiolic acid C3H2O2 | 207 | 1,114 |
| Methyl nitrate CH3NO3 | 676 | 4,835 |
| Lactaldehyde C3H6O2 | 1,366 | 10,103 |
Major publications
- A Scaled Hypersphere Search Method for the Topography of Reaction Pathways on the Potential Energy Surface. K. Ohno and S. Maeda Chem. Phys. Lett., 384(4-6), 277-282 (2004).
- Systematic Exploration of the Mechanism of Chemical Reactions: Global Reaction Route Mapping (GRRM) Strategy by the ADDF and AFIR Methods. Satoshi Maeda, Koichi Ohno, and Keiji Morokuma Phys. Chem. Chem. Phys., 15, 3683-3701 (2013).
- Global Mapping of Equilibrium and Transition Structures on Potential Energy Surfaces by the Scaled Hypersphere Search Method: Application to ab initio Surfaces of Formaldehyde and Propyne Molecules. S. Maeda and K. Ohno J. Phys. Chem. A 109(25), 5742-5753 (2005).
Visit developers' sites for more info
Non-functional specs
- Item
- Content
- Operating environment
-
- Hardware
x86_64 computer that works below - OS
Red Hat Enterprise Linux 8.x or AlmaLinux 8.x *1
Red Hat Enterprise Linux 9.x, AlmaLinux 9.x or Rocky Linux 9.x *1 - Required software
Gaussian16 or Gaussian09 *2 - Optional software
Gaussian03, Molpro, GAMESS, ORCA, Turbomole, SIESTA *3
- Hardware
- Included items
- Software, installation manual *4 *5
*1 Virtual environments (WSL2, VirtualBox, VMware) are not supported for license management reasons.
*2 Not included in this product. Please prepare in advance.
*3 Not included in this product. Please prepare in advance as needed. A general interface to insert ab initio programs besides these is contained in GRRM26.
*4 Please refer to AFIR manual site for the latest GRRM26 info.
*5 GRRM is a registered trademark of the Institute for Quantum Chemical Exploration.
Functional specs
Update functions
- Function
- GRRM20
- GRRM23
- GRRM26
- Reaction pathway search by AFIR method
- ○
- ○
- ○
- Kinetic simulation and kinetics-based navigation using RCMC
- ○
- ○
- ○
- Quantum chemistry-aided retrosynthetic analysis (QCaRA)
- ✕
- ○
- ○
- Utilities to easily implement user-developed tools
- ✕
- ○
- ○
- Optimized search performance for high-speed energy calculation programs (SEM / MLP)
- ✕
- ✕
- ○
- Explicit consideration of concentration effects in RCMC calculations
- ✕
- ✕
- ○
- RCMC under a Microcanonical Ensemble condition (f-RCMC)
- ✕
- ✕
- ○
- Improved adaptability to the virtual ligand method
- ✕
- ✕
- ○
See more
See more
GRRM26
GET IN TOUCH
GET IN TOUCH
For more information, please contact us.
We support users to set up and start GRRM26 (free of charge in standard).
For more in-depth support on how to use GRRM26 or how to interpret results, please consider our paid GRRM support service. Please contact us through our inquiry form.
Please refer to AFIR manual site for the latest manual.
-
Released "GRRM26"!!
July 17th, 2026